IDMP (Identification of Medicinal Product) is a set of five standards developed by ISO to enable the unique identification of a medicinal product using a standard language across their entire lifecycle – from development to authorisation and marketing.
This is ensured through consistent documentation and terminologies to describe the medicinal product and the exchange of product information between global regulators, manufacturers, suppliers, and distributors.
Unique identification helps to improve product and patient safety for medicinal products across a range of life sciences organisations, such as CROs, biopharma companies and global regulators.
The unique identification helps to improve product and patient safety for medicinal products across a range of life sciences organisations, such as CROs, biopharma companies and global regulators.
These five IDMP standards provide data elements and structures to uniquely identify and exchange information on:
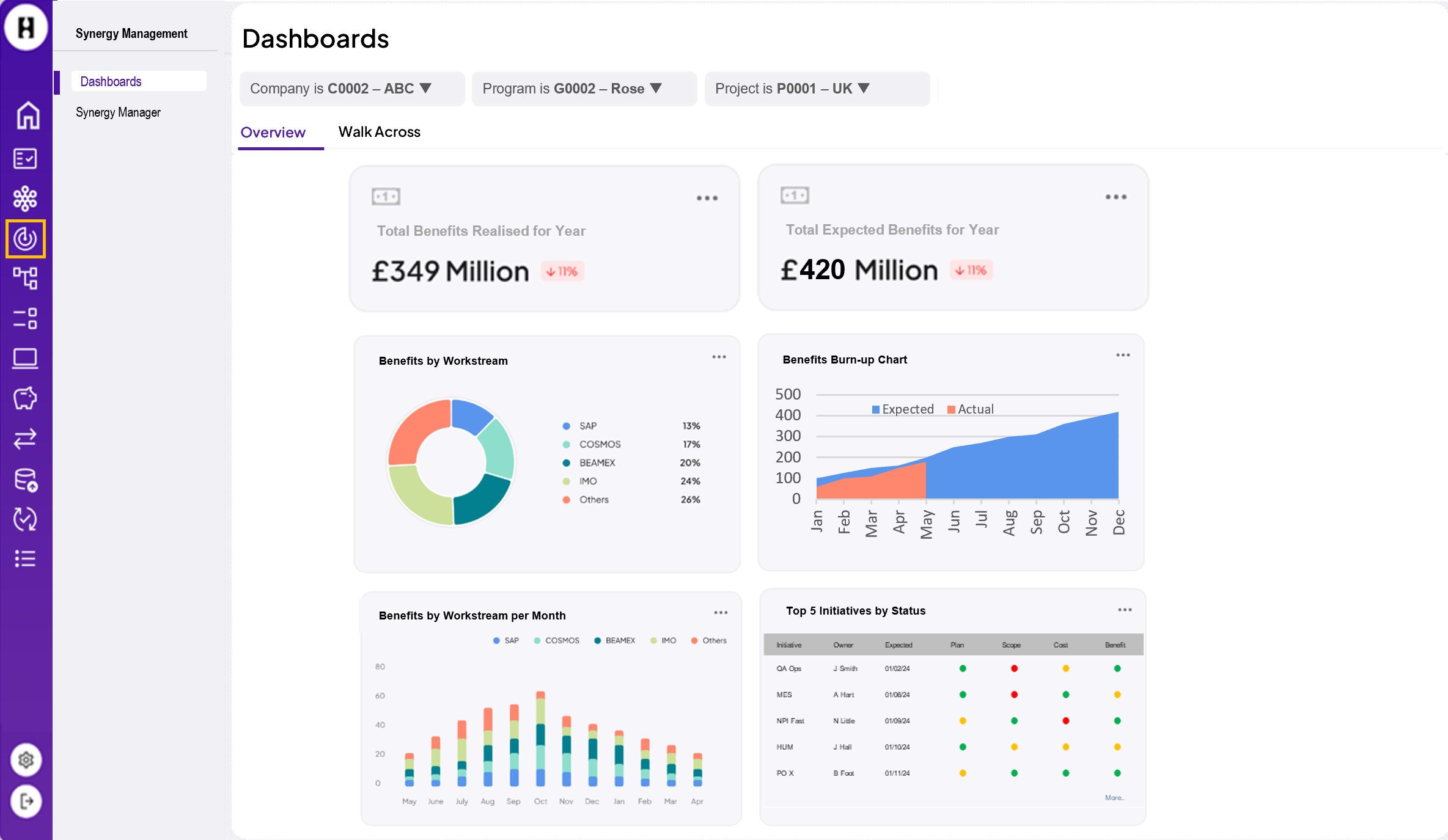
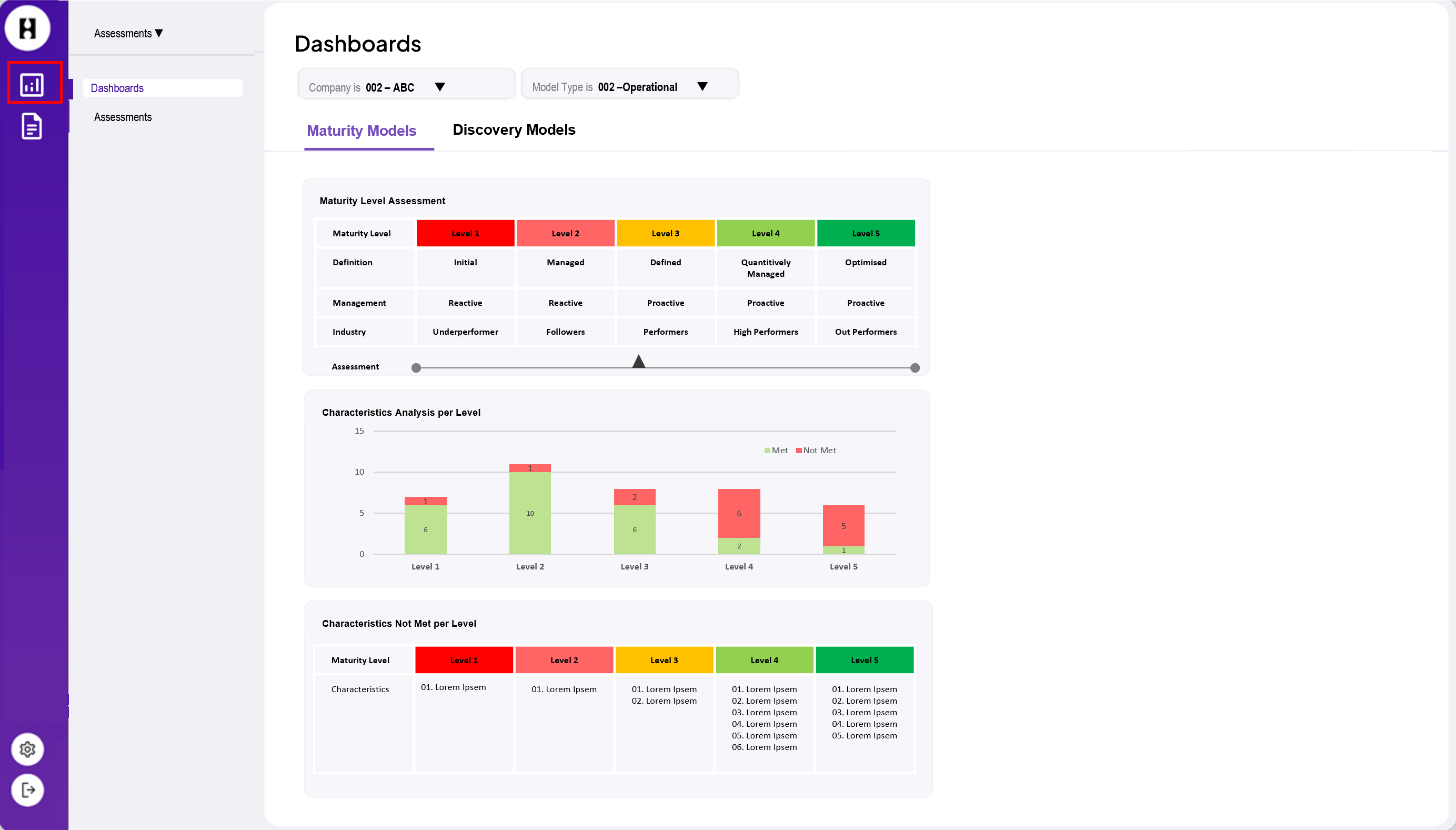
The EMA (European Medicines Agency) is in the process of implementing these standards in a phased program based on four domains of master data. This is called SPOR.
These data management services help to centralise the management of master data that complies with the ISO IDMP standards. Since July 2012, the submission and maintenance of data on authorised human medicines has already been compulsory. This has been based on a format called Extended EudraVigilance Product Report Message (xEVPRM), which will be mandatorily replaced by the ISO IDMP compatible format from 2023.
Implementing the ISO IDMP standards will simplify the exchange of information between regulatory authorities and healthcare communities, thus ensuring wide interoperability. They affect every aspect of life sciences organisations from discovery to post-marketing.
If the data is already present in xEVMPD system for marketed products, then this information will be migrated into the PMS (Product Management System). The product information will need to be quality controlled and enriched via an API submission to PMS prior to any submission to regulatory authorities.
According to the new IDMP submission system, when an application is made for marketing approval and for any subsequent variations via EU centralised procedure, all the unstructured data coming from different sources needs to be converted into a structured format according to the IDMP guidelines. This structured data is then submitted to the regulatory authorities through an interactive web form. This form generates a human readable PDF rendition as well as a machine-readable FHIR rendition (Fast Healthcare Interoperability Resources), which will be attached to the working documents folder of the application form (eCTD) sent to the regulatory authorities.
Marketing Authorisation Holders (MAHs) need to ensure their Regulatory Information Management (RIM) system is up to date at all times as most of the data for submission comes through this system.
MAHs need to check their IDMP readiness.
IDMP ensures consistent documentation and terminologies to describe the medicinal product and the exchange of product information between global regulators, manufacturers, suppliers, and distributors.
Here at Helixr, we work with our partners to ensure compliance through their product lifecycles. Over the coming months, we will be issuing further guidance as well as developing tools to assist with the preparation for IDMP compliance. Watch this space!